Hepatitis-C Drugs And A Remdesivir Metabolite As New Anti-Covid-19 Drugs: The Viral Protein NSP3 Emerges As A New Target.
(Posted on Friday, June 4, 2021)
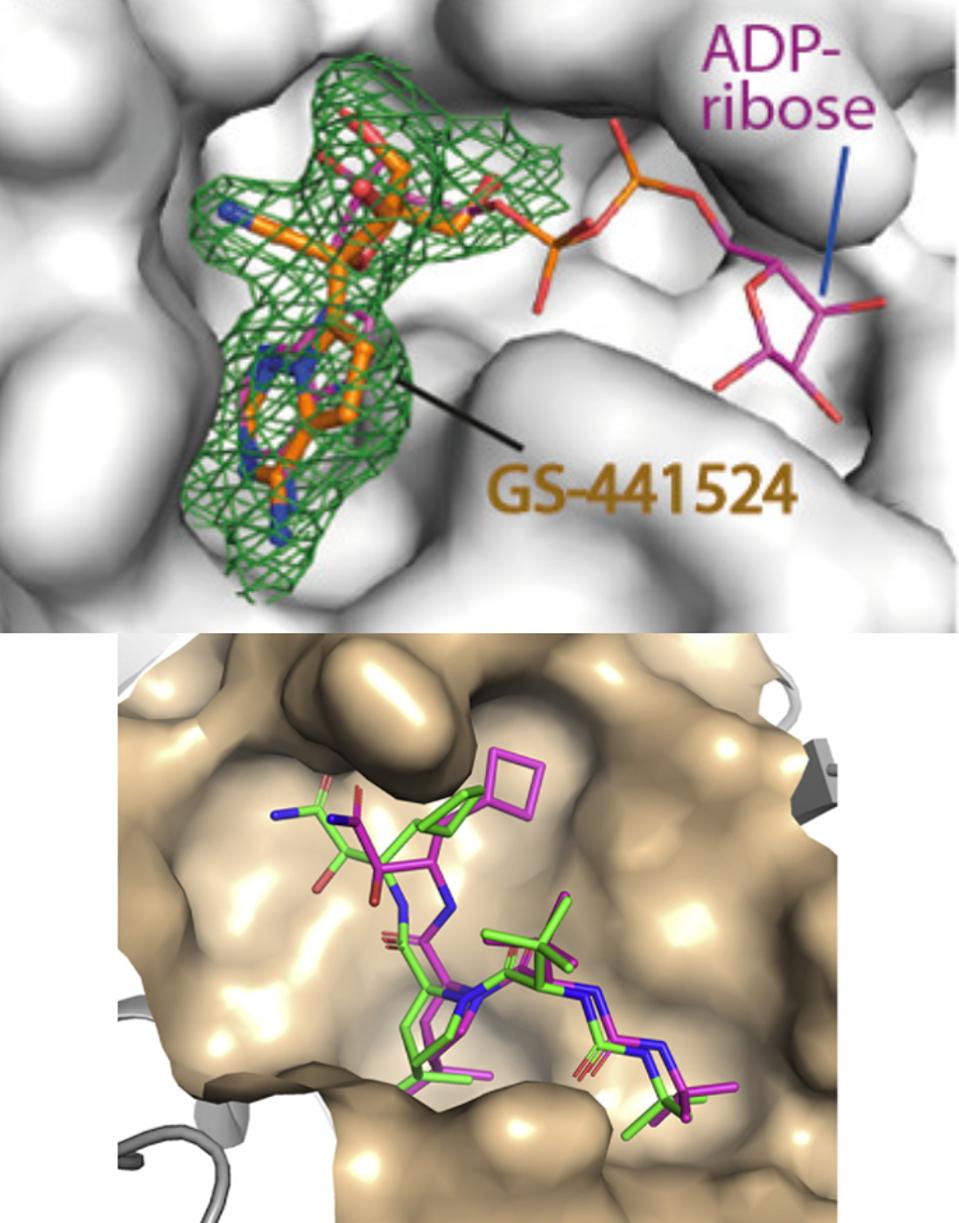
The GS-441524 Remdesivir metabolite alongside ADP-ribose; HCV drug BOC binding to the NSP5 protease binding pocket.
NI ET AL. // BAFNA ET AL.
In the search for new ways to use small molecule drugs to prevent and treat Covid-19 infections, a surprising synergy has emerged. Two drugs—Remdesivir metabolite GS-441524 and combination Hepatitis-C antivirals—both of which target NSP3 hold immediate promise for prevention and treatment of infection by SARS-CoV-2. Some of these drugs are highly synergistic, as much as tenfold, and show few adverse toxic effects. Here is the story of how they were discovered. It is also a surprising story about repurposing old drugs for new uses, and the discovery of an unlikely new target of anti-Covid-19 drugs, the viral protein NSP3.
We are now halfway through year two of the Covid-19 pandemic. The end is not yet in sight given the recent surge in India, the increase in infections in Latin American, and flare-ups in countries such as Japan, Taiwan, Singapore, Thailand, Vietnam, and Australia. Vaccines will be the backbone of Covid-19 control as evidenced by the positive results in Israel, the United Kingdom, and most recently the United States. However, the variability of protection offered by different vaccines over time and against viral variants warrants consideration for other means to prevent infections of the unvaccinated, for cases of vaccine breakthrough, and infection of those with underlying immune dysfunction.
I believe that the successful Covid-19 control strategy will be multi-modal, combining vaccines, anti-SARS-CoV-2 antibodies, and combinations of antiviral drugs. Our ongoing series outlines rapid progress in progress in anti-SARS-CoV-2 monoclonal antibodies and nanobodies. Here we begin a new series devoted to progress in the development of small molecule drugs designed to prevent and treat Covid-19.
Viral protein NSP3 as a target for antiviral drugs
The genome of SARS-CoV-2 (SARS-2) is both large and complex, encoding at least 30 distinct proteins. The proteins can be divided into three function groups: Those that specify the replication-transcription machinery necessary to assure copying of the genome and production of messenger RNAs from which all the viral proteins are made, the structural proteins comprising the nuclear core, the outer membrane, and the spike proteins of the infectious virus particle, and the accessory genes, those that specify functions vital for successful propagation in adult immunocompetent animals but dispensable for growth in cell culture (Figure 1A).
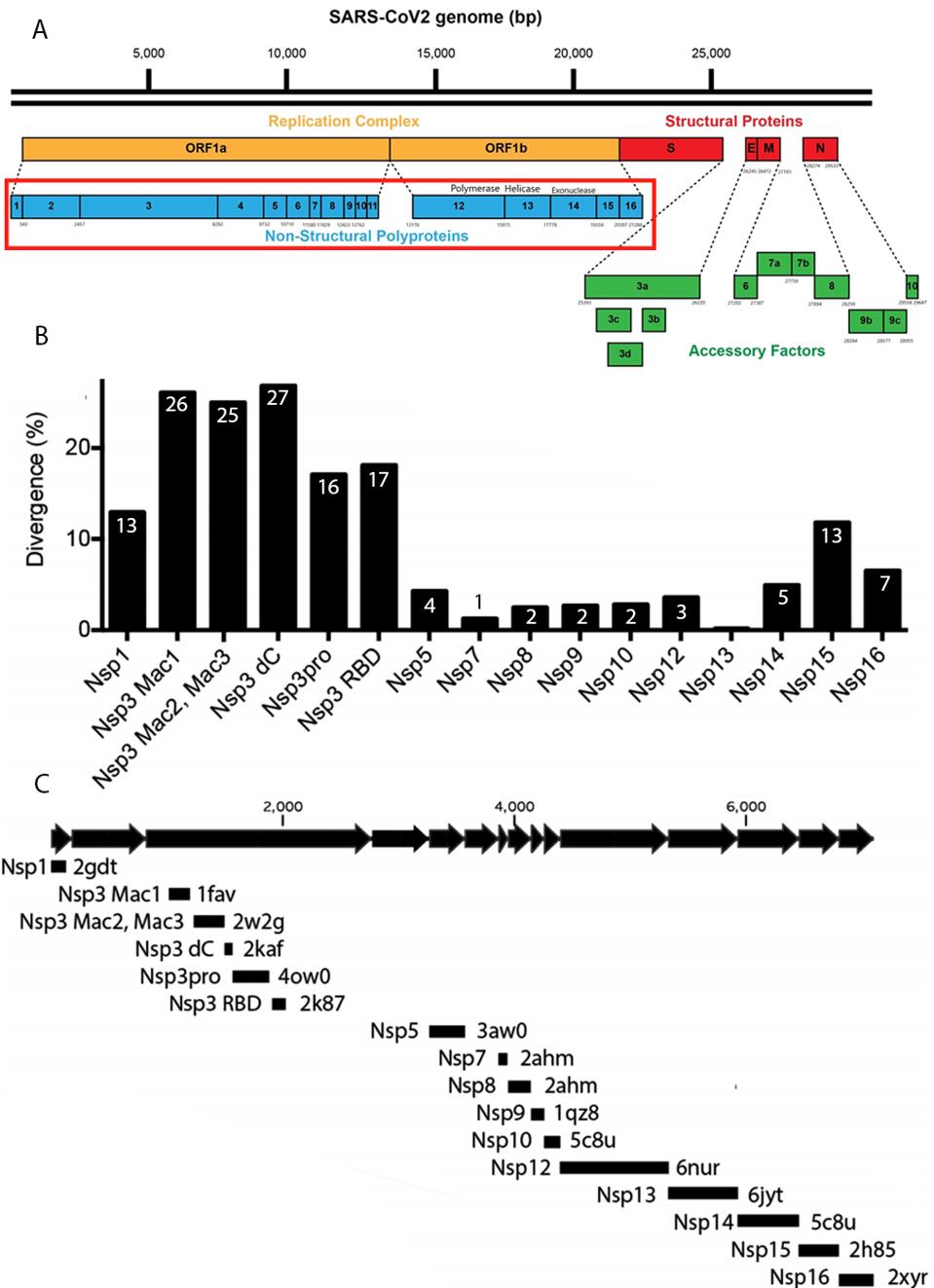
FIGURE 1: (A) SARS-CoV-2 genome. Orange indicates the SARS-CoV-2 replication complex; blue indicates the nonstructural proteins that comprise the replication complex; red indicates the structural proteins; green indicates the accessory factors. (B)
FRICK ET AL
The two long open reading frames at the 5’ end of the genome comprising roughly two-thirds of the entire genome specify the proteins designated NSP1-NSP16. NSP stands for non-structural proteins as they are not found in infectious viral particles. All of these proteins, NSP1-16, are essential for prolific replication in both cell culture and in animals. As such, they provide a wealth of targets for antiviral drug development.
In this series, I will enumerate the functions of each of these early essential genes and proteins and evaluate them as targets for antiviral drugs. We will begin with considering NSP3 (nonstructural protein 3) as a potential target.
NSP3 as a target for antiviral drug development
NSP3 is the largest and most complex of the SARS-2 proteins. The mature form of the NSP3 is almost 2000 amino acids long. It is synthesized as part of two much longer polypeptides, the product of the initial long open reading frame (orf1a), and from the orf1b, the orf1ab polypeptide. NSP3 frees itself in an autocatalytic process, along with NSP1 through NSP4 (Figure 2).
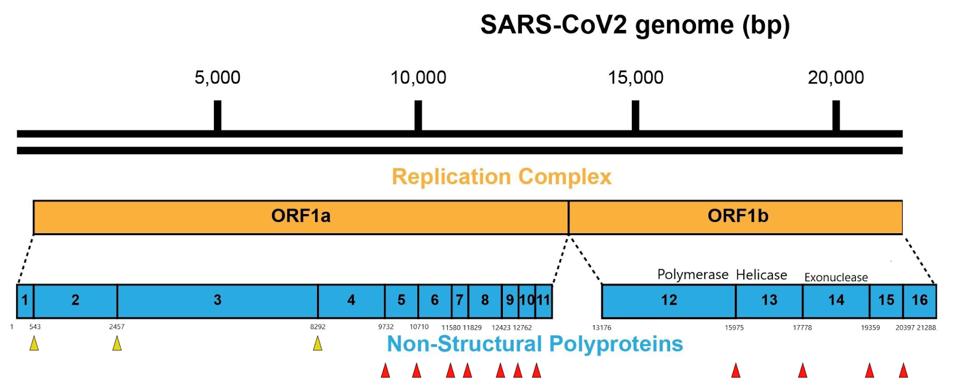
FIGURE 2: The NSP3 papain-like protease (PLpro) releases NSP1, NSP2, NSP3, and NSP4 during the processing of viral polyproteins, indicated by the yellow arrows. Red arrows indicate the NSPs released by the NSP5 main protease (Mpro).
ACCESS HEALTH INTERNATIONAL
The central role of NSP3 in early viral replication
All nidoviruses, of which the coronavirus are a subset, specify NSP3-like proteins, including SARS-CoV (SARS-1) the causative agent SARS. It is noteworthy that the greatest divergence of SARS-2 from SARS-1 extends over the entire orf1a open reading frame, including the region that encodes NSP3 (Figure 1B). The marked difference in this region of the genome points to an as yet unknown non-human precursor virus of SARS-2. The difference also suggests that the difference in biology—the long asymptomatic period, decreased pathogenicity, eventual pathogenesis, and late sequelae of SARS-2 as compared to SARS-1—might be in large measure determined by these differences in the orf1a proteins. Indeed, differences in pathogenicity of the porcine reproductive and respiratory syndrome virus are attributed to mutations in the orf1ab proteins of the NSP9 and NSP10 proteins.
Structural studies of the mature NSP3 reveal that the protein folds into seven discrete domains. Each domain species has one or more discrete functions. Table 1 summarizes what is known about the function of each of these domains. Also shown are the structural similarities of each domain with other cellular proteins of known function, aside from the two ends of the protein, which are relatively disordered.
The following figure shows the full crystallization structure of NSP3 (Figure 3).
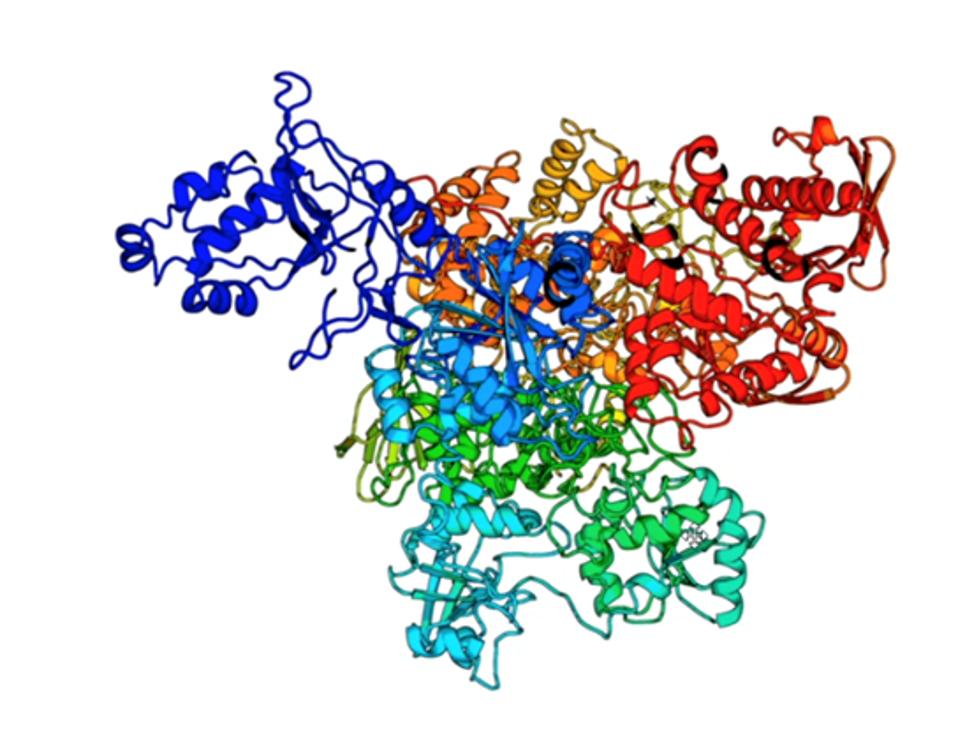
FIGURE 3: The NSP3 crystal structure.
UNIVERSITY OF MICHIGAN
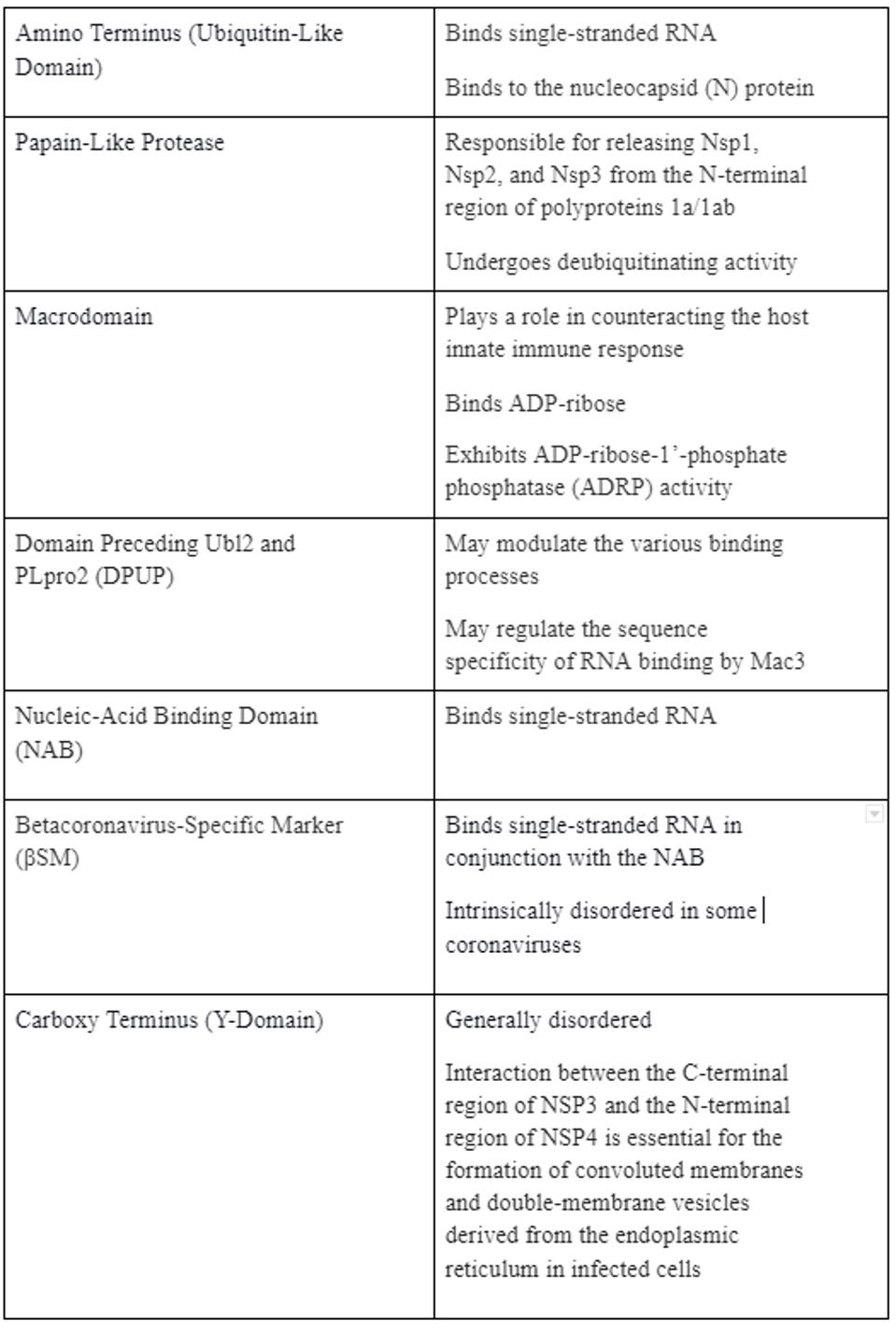
TABLE 1: Function of each domain
ACCESS HEALTH INTERNATIONAL
Many of the crystal structures of these domains have been determined independently, and each and every one of these domains may prove to be viable targets for small molecule drugs (Figure 1C).
The picture that emerges from the collection of these studies is that NSP3 is a linchpin of early functions required for virus replication. NSP3 is critical to:
1. The formation of the double-membrane vesicle within which viral replication and transcription occur (Figure 4).
2. Exit of the mRNAs from the double-membrane vesicle to the cytoplasm for translation of viral proteins via a highly structured pore .
3. Anchoring the replication complex to the inner surface of the double-membrane vesicle via interaction with both the membranes and the nucleoprotein (N) via tight association with the viral nucleoprotein (N) that binds viral RNA.
4. Protection of viral proteins from degradation. Modification of proteins by the addition of either ADP-ribose or ubiquitin marks the modified protein for degradation by cellular enzymes. One domain of the NSP3 removes ADP-ribose from modified viral proteins and others de-ubiquitinate marked proteins.
5. Proteolytic cleavage of the orf1a and orf1ab precursor polypeptides proteins to release the NSP1, NSP2, itself, and NSP4 of mutations that abrogate the NSP3 protease function are lethal to virus replication in cell culture and in animals.
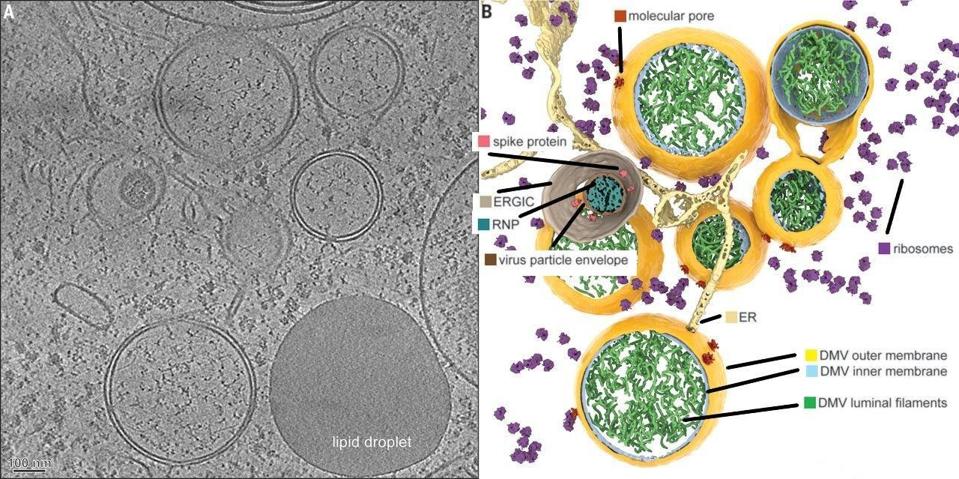
FIGURE 4: The SARS-CoV-2 double-membrane vesicle, which is primarily constructed by NSP3, NSP4, NSP5, and NSP6.
WOLFF ET AL.
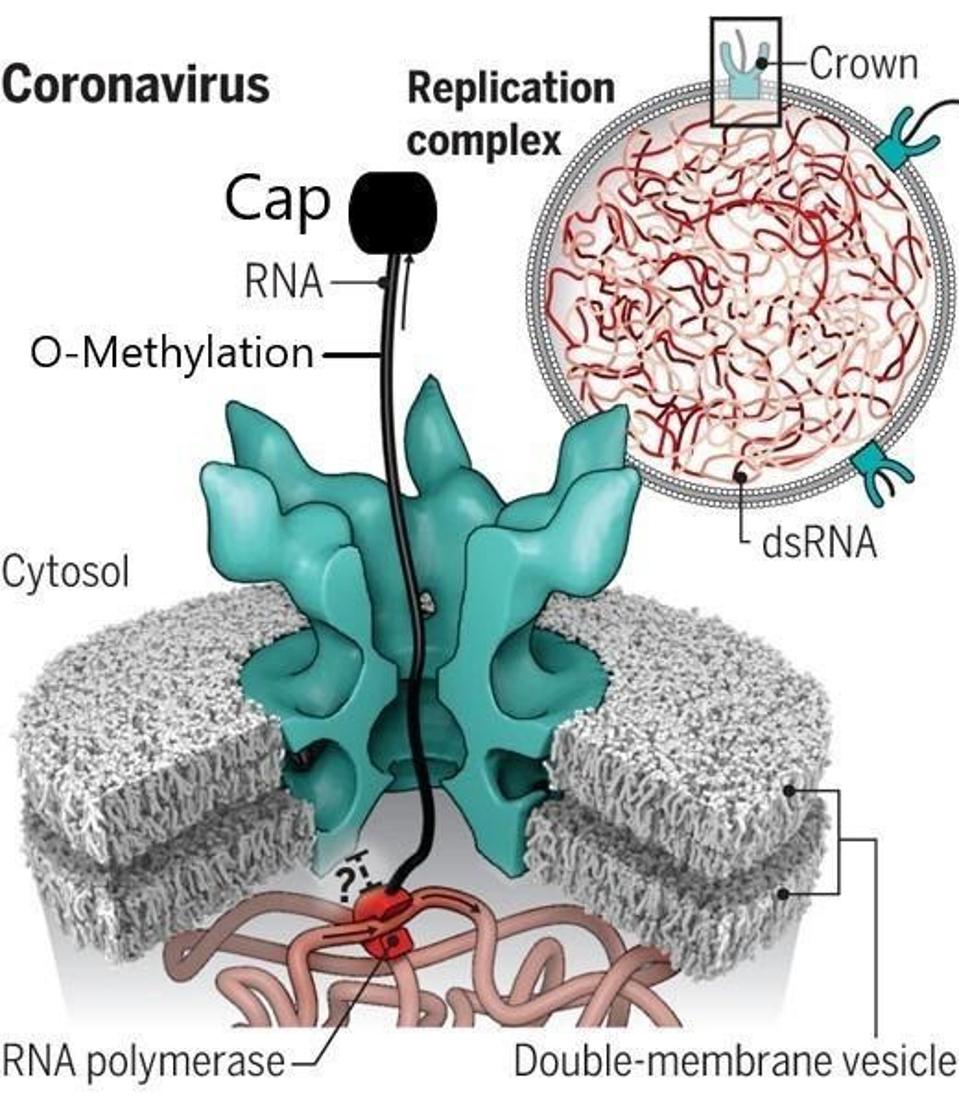
FIGURE 5: The pore created by NSP3.
UNCHWANIWALA ET AL.
High-resolution structures of the entire NSP3 protein as well as most of the individual domains have been determined by X-ray crystallography, cryo-electron microscopy, and nuclear magnetic resonance. The detailed structures facilitate computational docking with many thousands of potential drug candidates. Here, the focus falls on two specific domains, but we reiterate that there are many domains that could and should serve as potential targets for future antivirals.
Inhibitors of the NSP3 protease
A study analyzing hepatitis C antivirals assessed the ability of 10 available HCV protease inhibitors to suppress SARS-2 replication. Due to the structural similarity of the HCV protease to the SARS-2 NSP5 protease, the researchers hypothesized that the HCV drugs may also inhibit the NSP5 protease. Subsequent studies report three drugs, boceprevir, narlaprevir, and telaprevir, inhibit NSP5 protease proteolytic activity and bind into its active site.
Using virtual docking experiments, the researchers confirmed their suspicions, finding that all 10 of the HCV antivirals they tested can bind into the NSP5 protease binding cleft (Figure 6). Seven of the drugs inhibited both SARS-2 NSP5 protease activity and SARS-2 virus replication in Vero and/or human 293T cells. Surprisingly, they found that four of the 10 also inhibited NSP3 protease activity. Their encouraging finding was that HCV drugs that inhibit the NSP5 protease and/or the NSP3 protease can suppress SARS-2 virus replication.
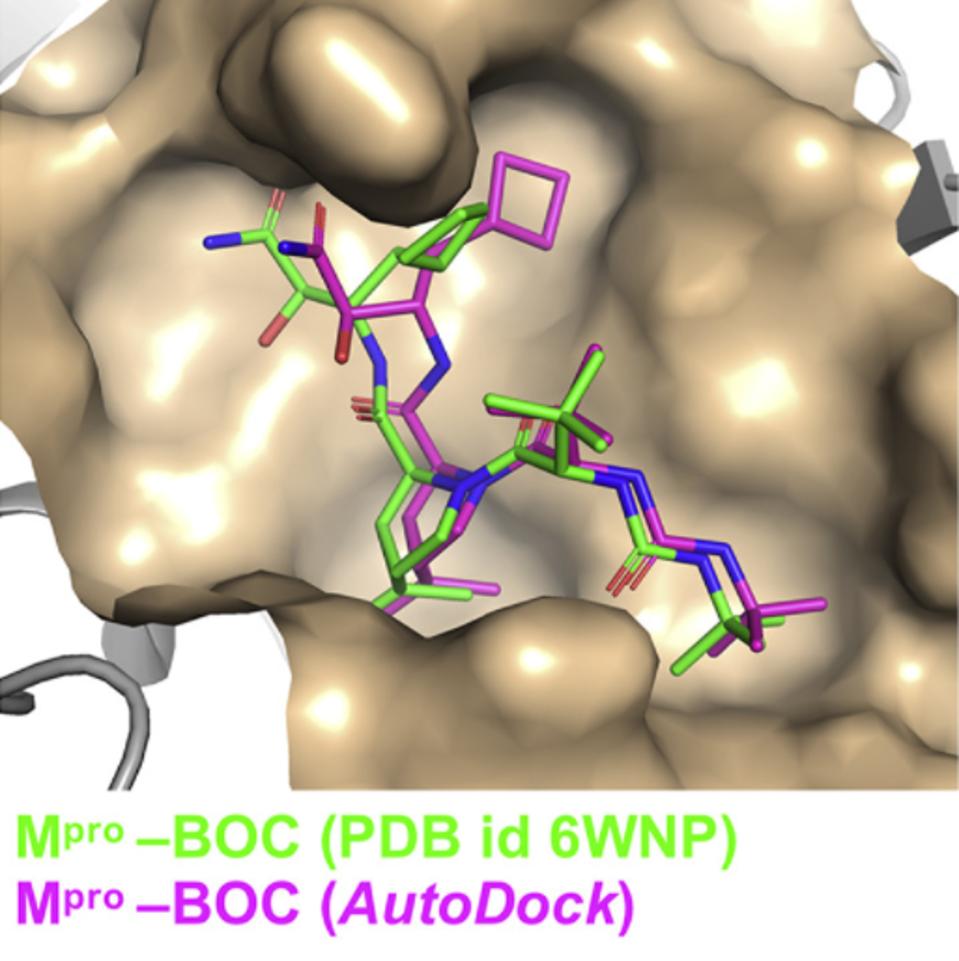
FIGURE 6: HCV antiviral BOC binding to the NSP5 protease binding pocket.
BAFNA ET AL.
While the active site of the NSP3 protease does not have much structural similarity with the HCV or NSP5 proteases, the researchers carried out virtual docking studies of these same 10 HCV drugs into the substrate-binding cleft of NSP3 protease. The known NSP5 protease inhibitor GRL0617 was used as a reference. They found that four drugs inhibited the NSP3 protease to similar levels as GRL0617. These proof-of-concept docking studies suggested that, surprisingly, some HCV protease inhibitors may bind in the substrate-binding clefts of both NSP3 and NSP5 proteases (Figure 7).
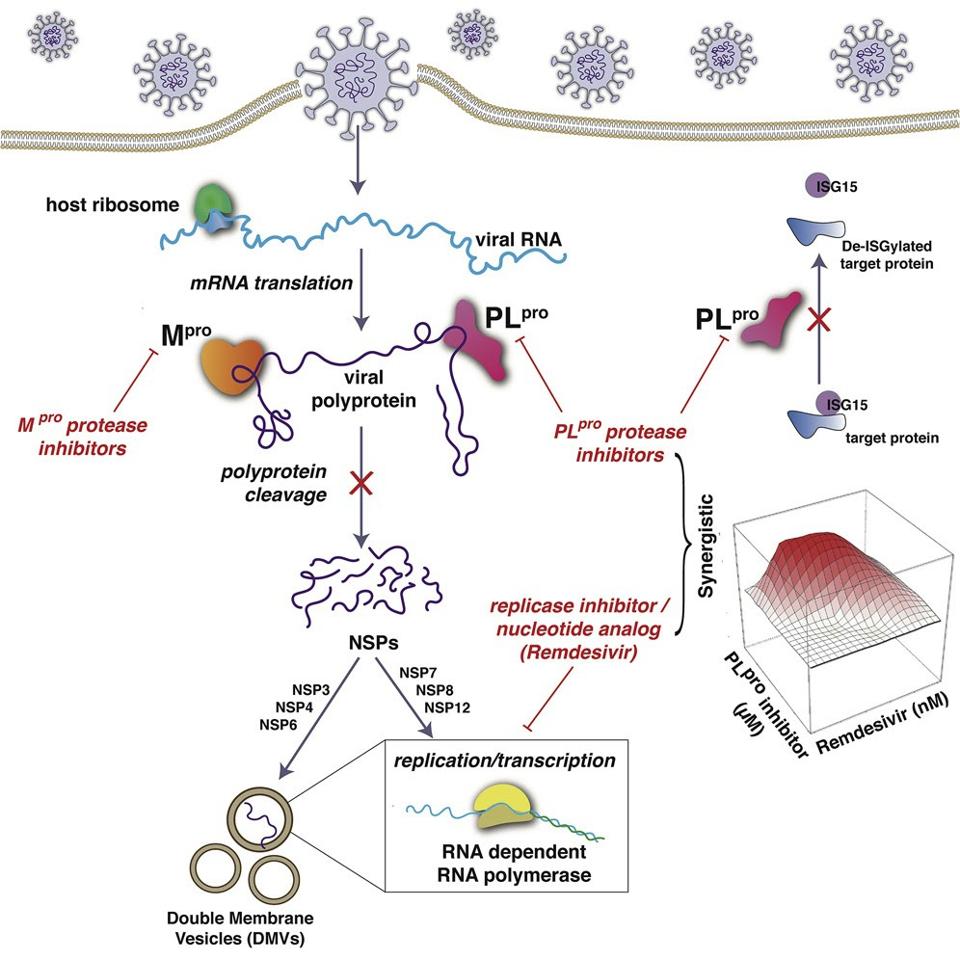
FIGURE 7: HCV antiviral papain-like protease inhibitors. ISG15 (interferon stimulated gene 15); PLpro (papain-like protease); mRNA (messenger RNA); Mpro (main protease).
BAFNA ET AL.
The researchers further demonstrated that these four drugs also acted synergistically with remdesivir to inhibit SARS-2 replication, thereby increasing remdesivir antiviral activity as much as 10-fold. In contrast, the HCV drugs BOC and NAR, which inhibit the NSP5 protease but not the NSP3 protease acted additively rather than synergistically with remdesivir. This suggested that the combination of an HCV protease inhibitor with an RNA polymerase inhibitor could potentially function as an antiviral against SARS-CoV-2.
They conclude by remarking that “more generally, our results strongly motivate further studies of the potential use of PLpro protease inhibitors in combination with RNA polymerase inhibitors as antivirals against SARS-CoV-2.”
Inhibitors of the NSP3 ADP-ribose phosphatase
In addition to the HCV drugs, there is another that targets the NSP3 macrodomain. The macrodomain and its ADP-ribose removal function are important for efficient viral replication, as viruses harboring inactive macrodomains exhibit reduced replication ability and pathogenicity and are sensitive to interferon pretreatment. ADP-ribose removal has also been linked to the function of viral proteins involved in viral replication or in immune antagonism.
The central role of the SARS-2 macrodomain in viral replication and immune responses suggests that this protein module may offer a target for combatting the disease. Some subtle structural differences between SARS-2 and other coronaviruses have been observed, suggesting that macrodomain may possess some degree of plasticity, opening a possibility for the binding of diverse small molecules, which researchers investigated in a study by Ni et al.
To understand the macrodomain plasticity, they crystallized the domain in an apo state. The atypical presence of five and three molecules in their asymmetric units suggested potential flexibility. Then, a structural comparison of the macrodomain’s chains revealed some differences in the residue conformation lining the ADP-ribose binding pocket. The researchers observed diverse conformations of residues in the phosphate-binding site. The flexibility of phenylalanine and isoleucine indicated the pocket may be open to binding a diverse set of ligands. Overall, the structural changes revealed the potential intrinsic plasticity of the macrodomain, especially within its ribose-phosphate binding pocket, indicating the potential binding of ligands.
This plasticity prompted the researchers to speculate the potential binding of antiviral nucleoside analogs. They selected a set of diverse antivirals, including abacavir, entecavir, acyclovir, gemcitabine, remdesivir, and remdesivir metabolite GS-441524, and tested the binding of these drugs using cocrystallization. Electron density maps revealed that the remdesivir metabolite GS-441524 was the only ligand that showed binding in the crystal structures. The interaction of this compound was rather unexpected because this metabolite and its active triphosphorylated form have been designed to target the NSP12 RNA-dependent RNA polymerase of several coronaviruses. Notably, an earlier study this year showed remdesivir metabolite GS-441524 potently inhibits SARS-2 infection in mouse models. Additionally, GS-441524 is also a proven treatment for cats with naturally occurring feline infectious peritonitis. The researchers’ structural comparison demonstrated that the binding mode of GS-441524 in the macrodomain highly resembled that of the adenosine moiety of ADP-ribose, indicating that the metabolite can bind to the macrodomain in place of ADP-ribose (Figure 8).
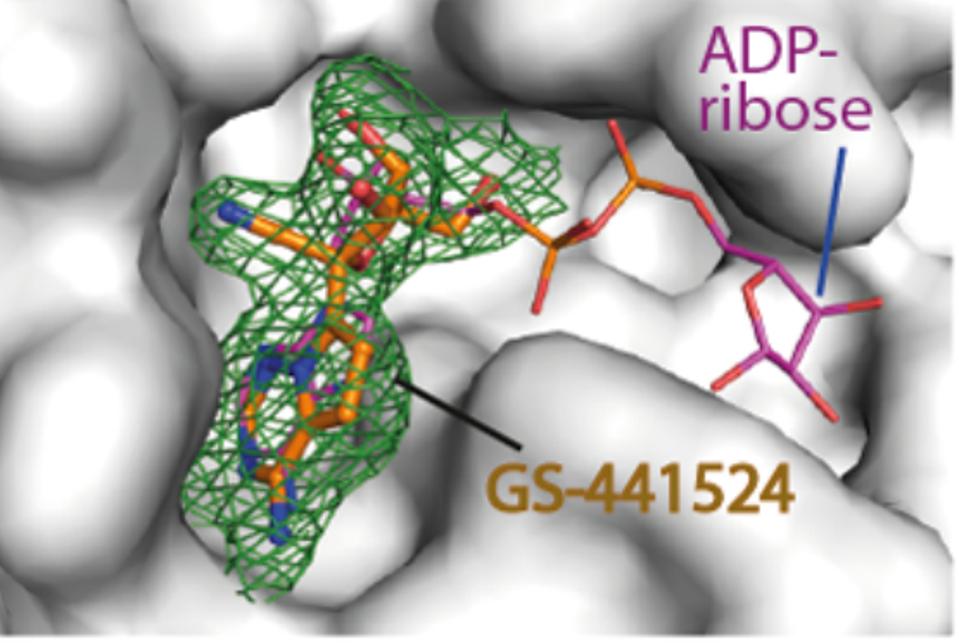
FIGURE 8: GS-441524 metabolite crystal structure alongside ADP-ribose.
NI ET AL.
The researchers additionally note that the NSP3 macrodomain is highly conserved, not only in SARS-2 variants but in SARS-1 and MERS-CoV as well. This opens the possibility of a broadly neutralizing antiviral. The issue with convalescent sera as a treatment to Covid-19 is that this virus adapts, developing mutations, and becoming resistant to older neutralizing antibodies. When we develop antivirals that target highly conserved areas, the potential escape of SARS-2 mutants is reduced as the target remains the same. This could ease the burden brought by highly infectious SARS-2 variants, as well as prepare us for potential coronavirus pandemics in the future.
The researchers conclude, “targeting the MD offers an attractive target for the development of antiviral agents against SARS-2 and other viruses.”
It is intriguing that these two classes of inhibitors target two different proteins. HCV antivirals inhibit both proteases and the Remdesivir metabolite inhibits ADP-ribose in the macrodomain, as well as the NSP12 polymerase through its active intermediate. These are powerful arguments for the combination of these two classes of antiviral to make an excellent prophylactic and therapeutic for prevention and treatment of Covid-19.
A combination therapy could be developed taking advantage of the synergistic effects of the HCV drugs and the potent neutralizing capabilities of the remdesivir metabolite. We already know HCV antivirals boost the potency of remdesivir, so it is possible that a similar effect could take place with the metabolite. As an added advantage, the protease, the ADP-ribose inhibitor, and remdesivir are available to be administered orally, meaning a combination could likely come in this form as well. These are just two of many antivirals that could be combined in a unilateral attack on NSP3. The protein has so many crucial targets; we must take advantage.
Read the full article on Forbes, (originally published on June 4, 2021).
Read Dr. Haseltine's latest piece with
![]()
