Whence And Whither Covid-19: The Many Faces Of Omicron
(Posted on Monday, February 28, 2022)
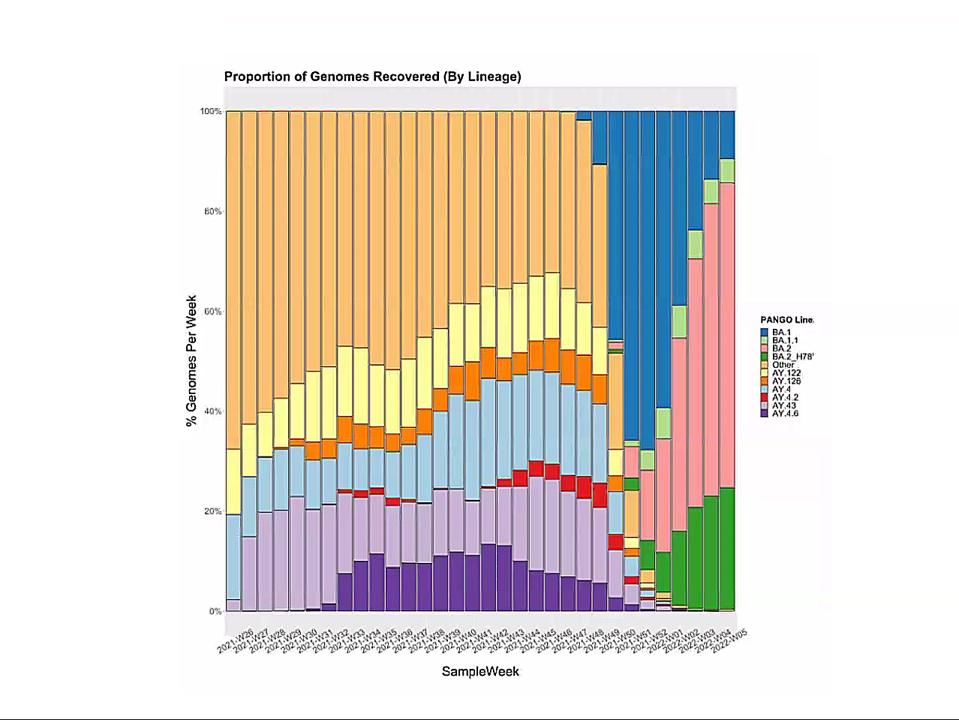
Emergence of Omicron sublineages. Blue is BA.1. Pink is BA.2. Green is BA.2 + H78Y.
DR. ERIC DING
What, without asking, hither hurried whence?
And, without asking, whither hurried hence!
Another and another Cup to drown
The Memory of this Impertinence!
Omar Khayyam / English version by Edward FitzGerald
We are all wondering what comes next for the Covid pandemic. Are we on the brink of the next wave of infections from an as yet unidentified variant? If so, will the new variant be more or less dangerous? Will our vaccines, those we have now and others that will arrive soon, protect us from infection, severe disease, and worse?
The short answer is that we do not know either whence or whither the SARS-CoV-2 pandemic. Yet, two years of experience provides some insight.
We know that new variants start small and multiply. They may begin almost anywhere, such as the UK, India, or South Africa, and spread globally, displacing all those that came before. The new wave may be intrinsically more lethal, as with Delta, or more contagious, as with Omicron.
In this context, I observe the Omicron family of SARS-CoV-2 variants. I call it a family as all Omicron variants stem from an as yet undiscovered parent (Figure 1). This cryptic parent gave birth to triplets siblings—Omicron BA.1, BA.2, and BA.3—all born at or near the same time. In the short span of three months, BA.1 spawned BA.1. 1, now carving a path through the US. BA.2 displaced BA.1 in Asia, Southern Africa, and European countries. BA.2 has a new offspring n BA.2 + Orf3a H78Y.
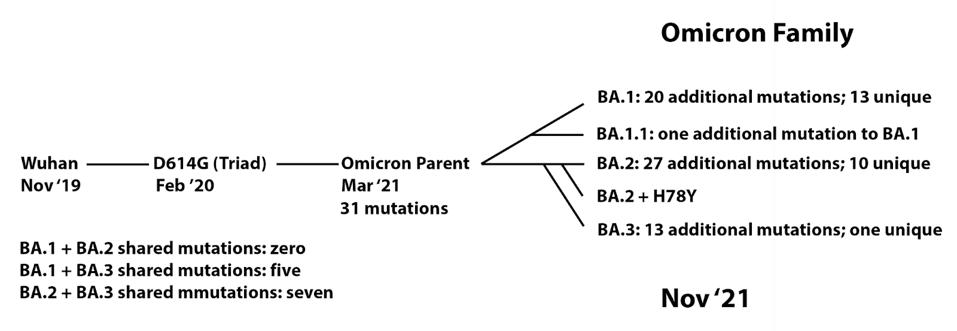
FIGURE 1: Proposed Omicron family tree, showing the development of mutations over time from the Wuhan strain to the Omicron sublineages.
ACCESS HEALTH INTERNATIONAL
Here, we focus on BA.2 + H78Y, the latest member of the Omicron family. We judge the inherent contagiousness of a new variant by its ability to replace prominent dominant viruses. Omicron BA.1 and BA1.1 displaced Delta as the dominant variant globally. BA.2 then displaced BA.1 and BA.1.1, where both branches occur. Now BA.2 + H78Y is replacing its parent BA.2 in Denmark. Despite our hopes that SARS-CoV-2 reached the limit of contagion with BA.1, both BA.2 and BA.2 + H78Y show such is not the case. BA.2 outcompetes BA.1, and BA.2 + H78Y outcompetes BA.2.
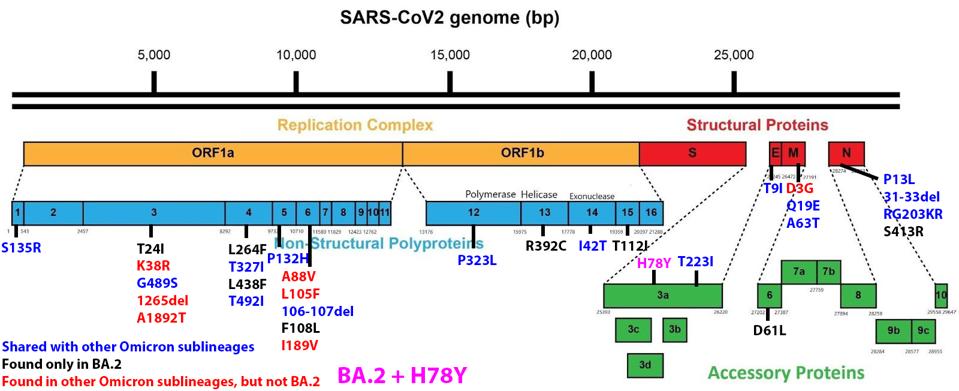
We have a powerful lens to view and perhaps understand what accounts for the increase in the replication rate of this newest variant—the sequence of the virus genomes. At first glance, the changes are surprisingly modest; one amino acid change in the Spike protein separates BA.1 from BA.1.1. Another single amino acid change in the Orf3a gene distinguishes BA.2 + H78Y from its parent BA.2 (Figure 2) (This analysis omits differences that might arise from mutations that do not alter viral proteins these called cis-acting mutations).
ACCESS HEALTH INTERNATIONAL
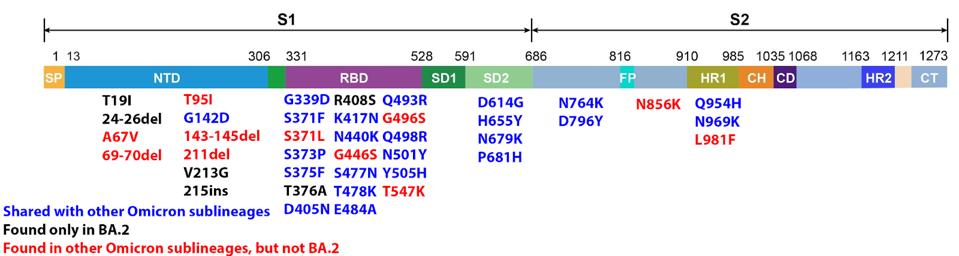
FIGURE 2: BA.2 + Orf3a H78Y mutation genome and Spike protein. Mutations in blue are shared between BA.2 and other Omicron sublineages. Mutations in black are unique to BA.2. Mutations in red are found in other Omicron sublineages, but not BA.2. The
ACCESS HEALTH INTERNATIONAL
BA.1.1 differs from BA.1 by changing the amino acid 346 from an arginine to a lysine (R346K). The change is relatively conservative, exchanging one positively-charged amino acid for another. The side chain of arginine is slightly longer than that of lysine. The best interpretations are that this subtle change shields the BA.1.1 Spike protein from recognizing some anti-spike neutralizing antibodies present in those who have recovered from an infection or are vaccinated. Viruses with enhanced immune escape capability will spread through such populations rapidly. This explanation fits the popular narrative comfortably that immune escape mediated by changes in the receptor-binding domain of the spike protein accounts for the replication success of new variants.
The same narrative does not account for the success of BA.2 + H78Y. The Spike protein of BA.2 + H78Y is identical to BA.2. Rather the difference lies in the accessory protein Orf3a, a change at position 78 of histidine for a tyrosine (H78Y). The shape of the two amino acids is similar, but they differ in charge and polarity. Histidine is polar and positively charged, whereas tyrosine is hydrophobic and uncharged (Figure 3).
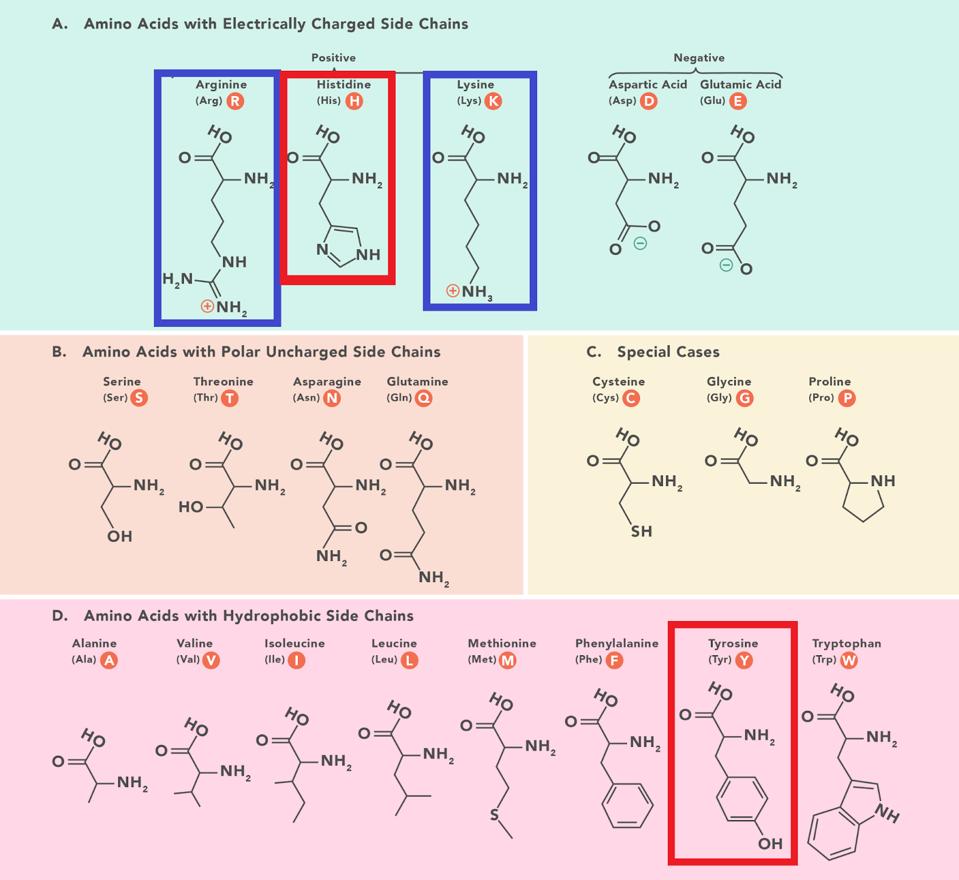
FIGURE 3: Amino acid structures with histidine and tyrosine highlighted. Amino acids are sorted based on two characteristics: hydrophobic vs. polar and charged vs. uncharged. Notice the similarity in chemical structure between histidine and tyrosine
DR. KAREN STEWARD // TECHNOLOGY NETWORKS
There are two general explanations to account for the difference in the two strains: immune escape or enhanced function, neither of which are mutually exclusive. Orf3a is immunogenic, recognized by both antibodies and T cells. A change in amino acid sequence might alter recognition by either antibodies, T cells, or both, providing the observed replication advantage.
Orf3a thwarts the innate immune response of infected cells. Innate immunity is the primary means our bodies protect us from novel pathogens. Innate immunity also limits the success of reinfection. Might the Orf3a H78Y mutation enhance suppression of the innate immune response? There is some evidence that subtle changes in accessory proteins account for the increased transmissibility of some SARS-Cov-2 variants. For example, Nevan Kroger and associates suggest that mutations that increase the production of Orf9b and Orf6, two proteins that also suppress the innate immune response, contribute to the replicative success of the Alpha variant.
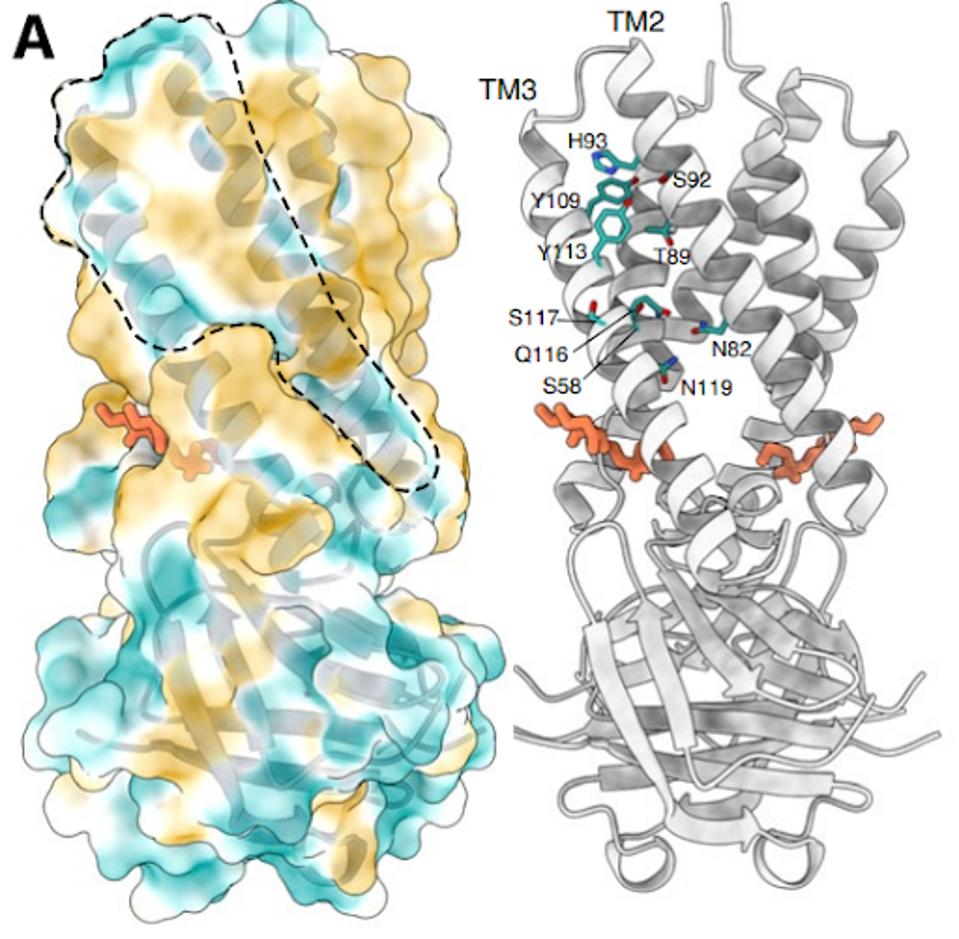
The structure of the Orf3a may offer a clue regarding the effect of the H78Y mutation on function. Two groups determined the three-dimensional structure of the protein. Orf3a is 275 amino acids long. The amino terminus of the monomer folds into three roughly parallel alpha helices that traverse the cell membrane. The carboxy-terminal constitutes the cytoplasmic domain (Figure 4a). The Orf3a monomers dimerize to form a pore in the membrane channel formed by six juxtaposed alpha helices (Figure 4b). The pore serves as a channel for transporting calcium ions (Ca+) and possibly others positively charged. We now add Orf3a to the known viral protein ion channels known as viporins (viral ion channel pore-forming proteins).
KERN ET AL.
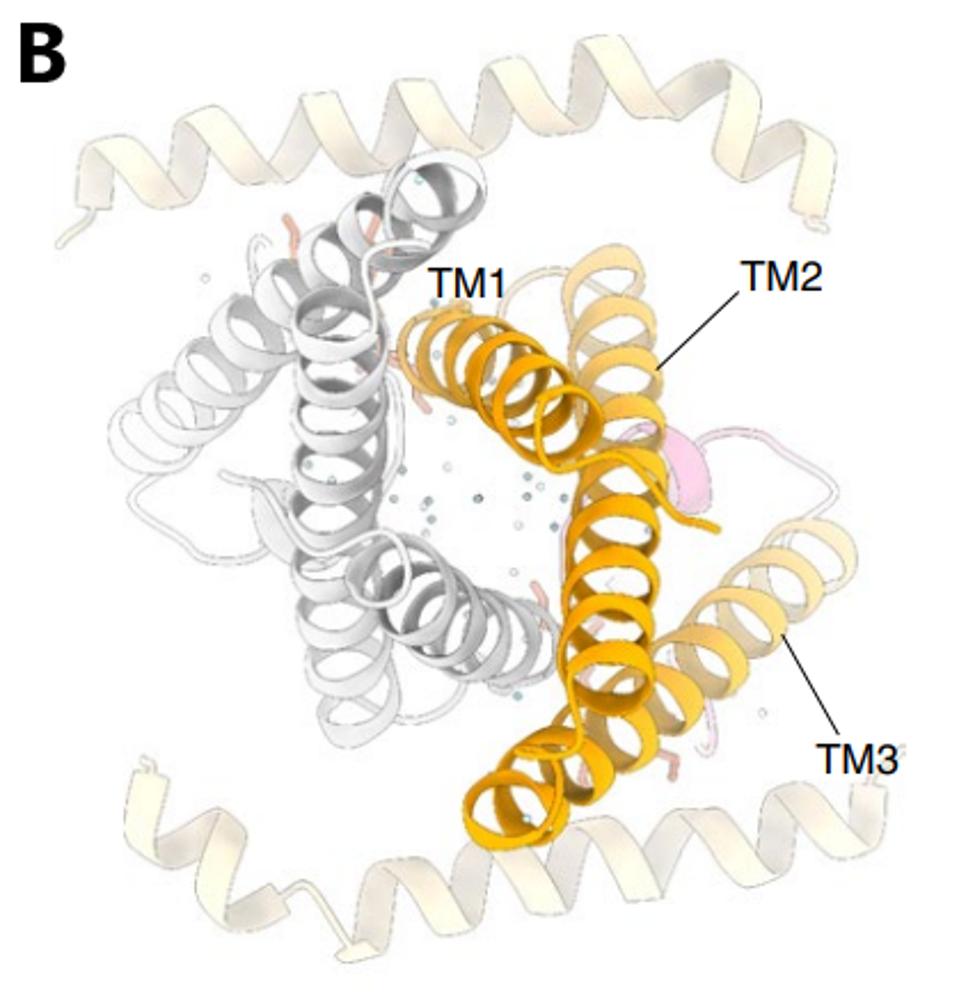
FIGURE 4: 3D rendering of the SARS-CoV-2 Orf3a protein. (A) The solvent-excluded surface of 3a colored from hydrophilic (dark cyan) to most hydrophobic (dark orange). A membrane-facing hydrophilic groove is outlined. Critical residues in the
KERN ET AL.
Amino acid 78 of Orf3a occupies a key site in the channel, facing the aqueous interior of the pore at what may be a choke point that determines the ease of passage of positively charged ions (Figure 5). A change from a positive to a neutral charge may facilitate calcium ion flux through the pore. Experiments to determine whether or not this is so are in progress (Brohawn, personal communication).
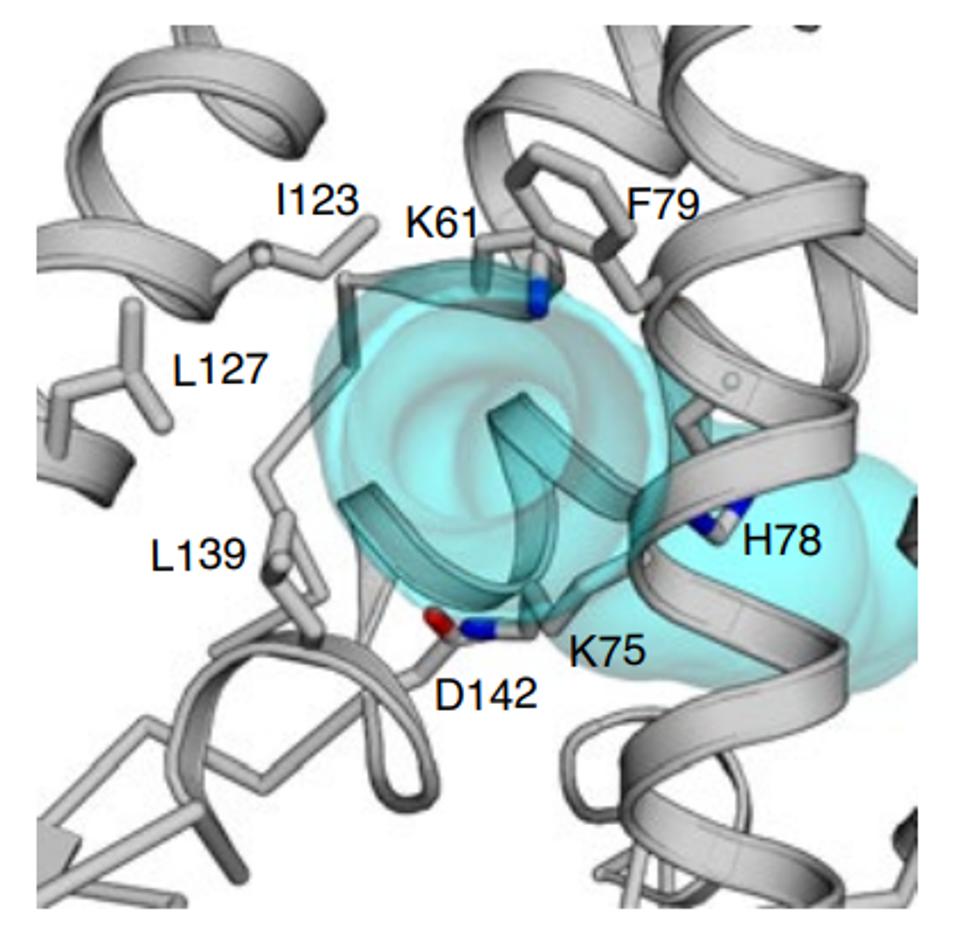
FIGURE 5: H78 as a potential choke point that determines the ease of passage of positively charged ions.
KERN ET AL.
Another outstanding question is the importance of the Orf3a ion channel activity to virus replication and the suppression of the innate immune response. The answer awaits the results of additional experiments. I note that single mutations in the ion channel of another viporin, the E protein of SARS-CoV-1, have a profound effect on the virulence of the virus in animal studies.
We note the H78Y in Orf3a has been sequenced 52,275 times in the GISAID database. While a majority of these sequences are recent, alongside the rise of the Omicron family of variants which began in November 2021, there are previous instances of H78Y in Orf3a. The mutation appeared in the United States in the short-lived “bluebird” variant that appeared in August 2020. In fact, throughout Europe and the United States, there are sequences of H78Y as early as March 2020. This may suggest that interacts with other mutations via epistasis, as the mutation has circulated for nearly two years without major implication until the rise of the Omicron family. We note that as of March 1st, Denmark represents the plurality of sequences with 19,723, followed by other European countries, for example England with 6,330 and Germany with 3,775. Some recent GISAID entries are also from the United States, suggesting that BA.2 + H78Y may have already arrived.
BA.2 + H78Y is a prime example of the significance and variability of SARS-CoV-2 variants. A single mutation in a non-Spike protein yields a potentially more transmissible virus that can outcompete its parental lineages. As the virus continues to spread and adapt, we anticipate more such examples appearing throughout the globe, extending the pandemic for the immediate future.
Read Dr. Haseltine's latest piece with
![]()
